CATEGORY
Clustering
SOURCE
RDKit
DESCRIPTION
Picks diverse subset of compounds using the MaxMinPicker algorithm as implemented in RDKit
INPUTS
A Dataset of Molecules to pick from.
Optional: a Dataset of Molecules that provides the seeds
See below for more details.
OUTPUTS
A Dataset of Molecules that comprises the newly picked molecules
OPTIONS
| Number to pick | The number of new molecules to pick | Positive integer |
| Threshold | Dissimilarity threshold (0.0 is identical) | Number between 0 and 1 |
| Fragment method | Strategy for selecting the largest fragment for multi component molecules | hac or mw |
| Output fragment | If multiple fragments then output the biggest, otherwise output the whole molecule | true or false |
| Descriptor | The molecular descriptor (fingerprint) to use to calculate the Tanimoto distance | maccs, morgan2, morgan3 |
ADDITIONAL INFO
Background
MaxMinPicker is an efficient algorithm for picking a optimal subset of diverse compounds from a candidate pool. The algorithm is described in Ashton, M. et. al. (doi 10.1002/qsar.200290002) and an improved RDKit implementation was described by Roger Sayle at the 2017 RDKit user group meeting. The Squonk implementation is based on that improved algorithm.
Squonk also has the RDKit Diverse Subset Picker cell (based on Butina clustering) that allows to pick a diverse subset and to optimise this for a particular property (e.g. logP) but this approach requires the whole distance matrix to be generated which limits scaelability (1000 structures is OK, 10,000 is not). In contrast the MaxMinPicker generates distances on demand and so is much more scaleable.
Variants
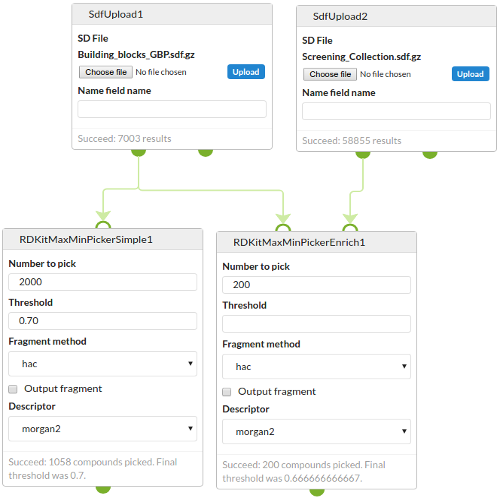
The MaxMinPicker cell comes in 2 variants:
RDKitMaxMinPickerSimple
This is for picking a diverse subset from a single pool of molecules. It has one input, the pool of molecules to pick from, and one output, the molecules that have been picked. It is broadly analogous to the RDKit Diverse Subset Picker cell but scales much better, though does not allow for optimisation of a molecular property.
RDKitMaxMinPickerEnrich
This is for picking a diverse subset from a single pool of molecules given a set of seed molecules. It has two inputs, one for the pool of molecules to pick from (named input) and, and another for the seed molecules (named seeds). It has one output, the molecules that have been picked from the pool. It is designed for scenarios like ‘given my existing compound collection give the the n most diverse molecules from a vendor catalog’.
Options
Whilst the inputs of the two variants are different the options are identical.
The Number to pick and Threshold parameters determine the picking criteria. One or both must be specified. The Number to pick parameter is used to terminate the picker once that many molecules have been picked. The Threshold parameter is used to terminate the picker once there are no more molecule that distant to any that have been picked (it is a dissimilarity measure), and as such can be used to allow to pick until the chemical space is saturated at that threshold. If both Number to pick and Threshold are specified then the picker terminates as soon and the first of them hits the limit.
Generating descriptors and distances only really makes sense for discrete molecules not mixtures. As salted forms and other types of mixture are common in molecule sets the MaxMinPicker provides options for how to generate the fragment to use for the picking. It does this by providing the Fragment method option that tells the picker how to determine the largest fragment where there are multiple fragments. It has 2 options, hac (heavy atom count) and mw (molecular weight). Where a molecule has multiple fragments the biggest according to hac or mw will be chosen and used to generate the descriptors. The Output fragment option specifies whether the picked molecules that are output should be the largest fragment or the entire molecule.
The final option is the Descriptor to use for the Tanimoto distance between molecules. Default is morgan2, but you can also use morgan3 or maccs keys.
Examples
The following screenshot shows both variants in use.