CATEGORY
3D
SOURCE
Commercial Open Source
DESCRIPTION
Ligand docking using rDock. There are two versions of rDock docking:
The original one named rDock docking where the receptor configuration is supplied as a zip file containing the receptor, the cavity definition and the docking parameters. This allows to define the docking configuration in full, but requires some knowledge of how to do this preparation (or have a zip file that someone has prepared for you).
An alternative named rDock filtering where the cavity definition is part of the cell execution based on the reference ligand that is specified as one of the inputs, which avoids the need to generate this, but relies on hard-coded parameters for the cavity definition and most of the docking parameters. Hence this is simpler, but less flexible. This variant also allows to filter the docked poses based on their normalised docking score relative to the reference ligand.
INPUTS
For rDock docking:
- A Dataset of Molecules with the ligands to dock
- A zip file with the docking configuration (see below)
For rDock filtering:
- A Dataset of Molecules with the prepared ligands to dock
- A reference ligand in molfile or SDF format
- The prepared receptor in mol2 format
OUTPUTS
- A Dataset of Molecules with the docked poses
OPTIONS
| Number of dockings | The number of docking runs for each ligand. Each run will generate a different pose. |
| Top scoring poses | The number of top scoring poses to output. |
| Score filter | (only for rDock docking) Optional score above which poses are not output. The score is an approximation to the binding energy, and will typically be a negative number like -23.1 with stronger binding having LARGER negative numbers. You need to study the scores against a particular target to get an understanding of a sensible filter to use. |
| Normalised score filter | (only for rDock docking) Optional normalised score above which poses are not output. The normalised score is the score divided by the number of heavy atoms in the ligand, and takes the size of the ligand better into account. |
| Score threshold | (only for rDock filtering) Normalised score Filter threshold (relative to normalised score for the the reference ligand). Larger negative scores are better so a threshold of zero results in only retaining poses with scores better than the reference ligand, a negative threshold is more stringent (poses x better than the reference ligand) and a positive threshold includes poses that much worse than the reference ligand. |
ADDITIONAL INFO
Introduction
rDock is a molecular docking program, originally created by RiboTargets, which became Vernalis. It was open sourced in 2012. For more information on rDock look here.
Inputs
Key to using the rDock docking cell is providing the docking configuration as a zip file. This file must contain the following:
- receptor.mol2 - the receptor structure in mol2 format
- receptor.prm - the rDock parameters file
- receptor.as - the cavity definition prepared using the rdock tools e.g. the rbcavity program
A good way of doing this is the Docking Validation project that Informatics Matters has created as an upstream project for Squonk. In particular there is an example for DHFR that you can follow that can be found here.
To generate the receptor structure you may need a third party program like MOE or Maestro, but we are hoping to provide a freely usable alternative in the future. To generate the parameters file and the cavity definition file see the rDock documentation. For information about Nextflow see nextflow.io. An example zip file is attached to this page.
The zip file can be uploaded using the ZipFileUpload cell.
For the rDock filtering cell you need to specify three individual inputs as described above.
Performing docking
Docking comprises 3 key steps
- Only for rDock docking cell: Preparing the docking configuration. Currently that is done outside of Squonk and the files packed up into a zip file. This is a complex process to be performed by an expert. Once done the zip file is uploaded using the ZipFileUpload cell.
- Only for rDock filtering cell: provide the receptor in mol2 format and the reference ligand in molfile format as inputs.
- Preparing the ligands. At the minimum 3D structures are needed, but you might also want to perform other operations such as enumerating stereoisomers and tautomers, and adjusting the charge state. Some prototype cells for these operations exist (e.g. Molecule Enumerator cell), but more work is expected here.
- Performing the docking using this cell. The output is the docker poses. It is expected that this is a relatively non-expert step and can be done by most users.
This is a minimal example of running a docking experiment with the rDock docking cell:
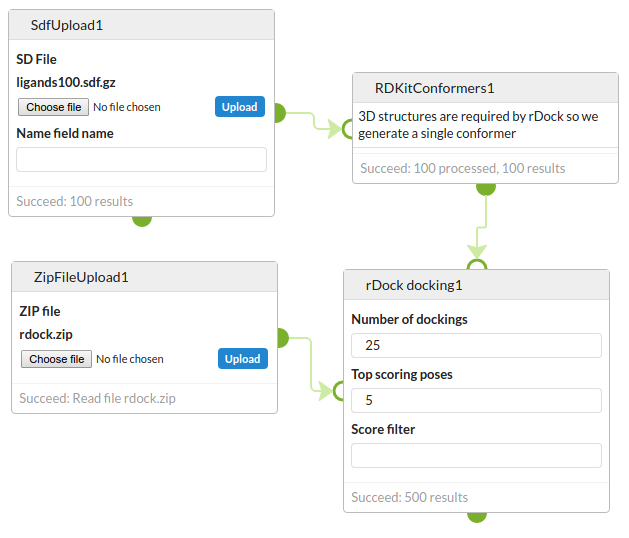
Typically you might view the output in the NGLViewer cell.
Future plans
We plan much more for this cell, and related cells. Some examples:
- More control over docking options
- Tethered docking
- Similar cells for other docking programs