CATEGORY
Molecular Screening
SOURCE
Other
DESCRIPTION
Alignment of 3D structures
INPUTS
A dataset of molecules containing conformers of structures. A field must identify the source molecule.
OUTPUTS
A dataset of molecules comprising the assemblies that are generated
OPTIONS
| Number of assemblies | How many assemblies to generate |
| Parent structure field | The name of the field that identifies the structure that each conformer corresponds to |
ADDITIONAL INFO
MolAlign is a set of programs from Ling Chan (slchan.bc@gmail.com). These have been made available to Squonk to allow feedback to be obtained.
Information about MolAlign can be found in this publication: DOI 10.1007/s10822-017-0023-8. Using MolAlign may incur costs in the future. Please contact Ling Chan if you are interested in using MolAlign outside Squonk.
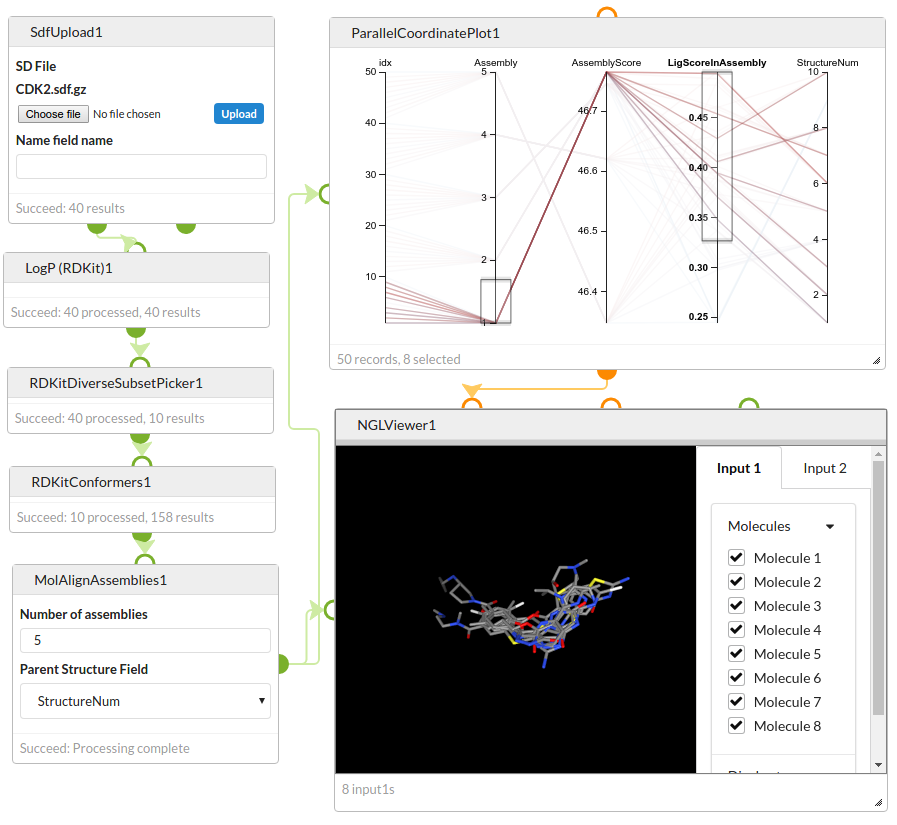
This variant of MolAlign generates optimal 3D overlays of the input structures. For each input structure a number of conformers must be provided. MolAlign creates a number of “assemblies”, each assembly containing a single conformer of each input structure with the conformers oriented for an optimum global alignment. Assembly 1 contains the best alignment, assembly 2 the second best etc.
The results contain the conformers in each assembly. The conformer is rotated and translated according to the alignment, but the internal geometry of the conformer is not modified. The same conformer may appear in multiple assemblies. The following fields are included:
| Field name | Description |
|---|---|
| Assembly | The assembly number. Number 1 is the assembly with the best alignment. |
| AssemblyScore | An alignment score for the assembly as a whole. Assembly 1 has the highest score. All members of the same assembly will have the same value. |
| LigScoreInAssembly | A normalised alignment score for each structure in the assembly. This is a number that will usually be between 1 (good alignment) adn 0 (bad alignment). This allows to identify structures that do not overlay well, and could be candidates for excluding from the alignment. |
| StructureNum | The ID of the input structure (corresponding to the value of the field specified by the Parent structure field options). |
As such it is suitable for cases where you have a set of know active compounds, but do not have crystal structures to provide you with 3D information. The assemblies may approximate to the bound conformations of the ligands on the principle that they must share a common 3D shape (pharmacophore).
MolAlign is fairly computationally demanding, and this increases dramatically with the number of structures and conformers of those structures. We find that computing assemblies for around 25 conformers of 10 structures completes fairly quickly. Going beyond those limits may result in greatly increased execution times. Please start with small numbers and be sensible as you increase this.
Tip: you can generate conformers using the RDKitConformers cell
Tip: you can generate a diverse subset from a larger number of input structures using the RDKitDiverseSubsetPicker cell.
Tip: you can visualise the results using the ParallelCoordinatePlot and NGLViewer cells.